


Warning¶
The Blue Brain Project concluded in December 2024, so development has ceased under the BlueBrain GitHub organization. Future development will take place at: https://github.com/openbraininstitute/morphology-workflows
Morphology Workflows¶
This project contains several workflows for processing morphologies:
Initialize: create the initial configuration files that the user can then update according to its needs.
Fetch: download morphologies from online database (like Allen, NeuroMorpho or MouseLight).
Placeholders: compute the placeholders for a given region and mtype set.
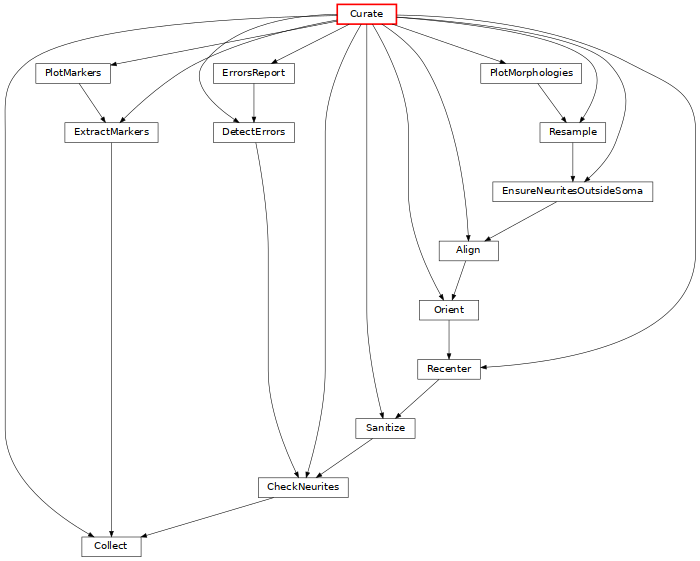
Curate: from raw morphologies, ensures that morphologies can be used with the rest of BBP codes (click on the image for more details).
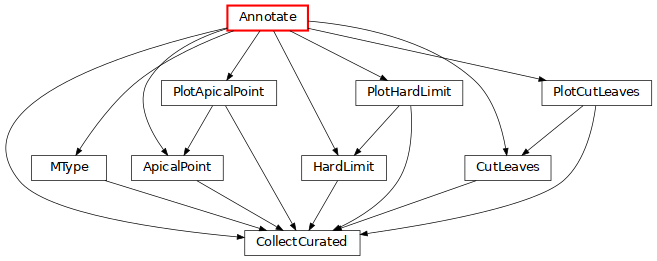
Annotate: create various annotations on morphologies needed by specific BBP codes (click on the image for more details).
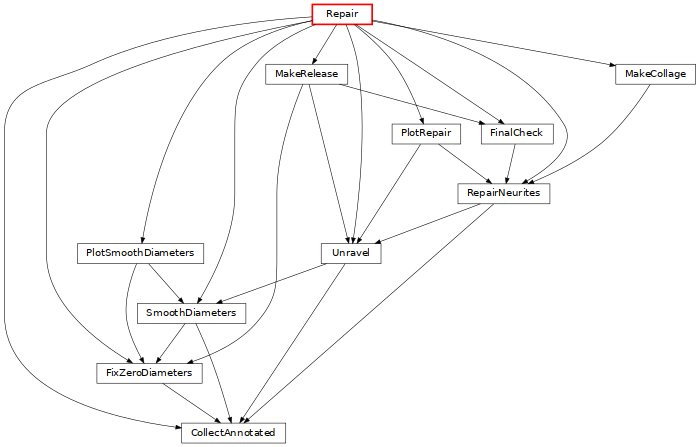
Repair: process morphologies to correct for artifacts of in-vitro reconstruction (click on the image for more details).
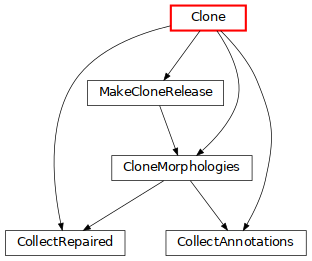
Clone: clone morphologies and apply some variability (click on the image for more details).
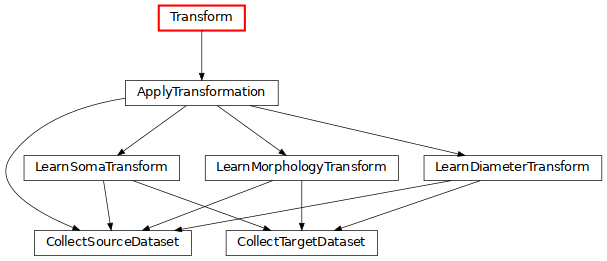
Transform: transform morphologies by rescaling the soma, the sections and their diameters (click on the image for more details).
In a nutshell, the user provides a list of morphologies in a .csv file, with their names and
paths and a luigi.cfg configuration file. Each workflow is run independently and creates an
output folder, with one subfolder per task. In each, there will be a report.csv and a data
folder containing the output files of the task if any. In the report.csv file, columns contain
paths to these files, additional information, error messages if the task failed on that
morphologies, as well as a flag is_valid, used in subsequent tasks to filter valid morphologies.
At the end of each workflow, another report.csv file is created, with the main output columns of
each tasks, and a report.pdf containing a human readable summary of the result of the workflow.
Usually, the user should run the Curate workflow, then the Annotate workflow and finally the
Repair workflow.
The complete documentation can be found here:
stable: https://morphology-workflows.readthedocs.io/en/stable/
latest: https://morphology-workflows.readthedocs.io/en/latest/
Installation¶
This should be installed using pip:
pip install morphology-workflows
Usage¶
Create inputs for the workflows using the Initialize workflow¶
Usually, user should create a new project folder using this workflow. It will create the files required to run the other workflows. For example, the command:
morphology-workflows Initialize --source-database NeuroMorpho
will create the inputs to download the morphologies from the NeuroMorpho database and the run the other workflows on these morphologies.
If the user wants to provide arbitrary morphologies to the Curate / Annotate / Repair workflows, the following command can be used:
morphology-workflows Initialize --input-dir <path-to-the-morphologies>
in order to create the dataset required by these workflows.
Once the Initialize workflows is complete, the user can update the outputs according to its needs (see the next sections for details about each workflow).
Create inputs for the Fetch workflow¶
This workflow helps fetching morphologies from online databases. This workflow only needs a configuration file, which depends on the source from which the morphologies are fetched.
The possible sources are:
NeuroMorpho
MouseLight
Allen
For each of them, the configuration file should be a JSON file containing a list of objects like the following examples:
NeuroMorpho:
[ { "species": "mouse", "brain_region": "neocortex", "cell_type": "interneuron", "nb_morphologies": 10 } ]
MouseLight:
[ { "brain_region": "neocortex", "nb_morphologies": 10, "seed": 0 } ]
Allen:
[ { "species": "Mus musculus", "brain_region": "VISli", "cell_type": "pyramidal", "nb_morphologies": 10, "seed": 0 } ]
In these examples, the seed attribute is optional and is only used to sample which morphologies
are fetched among those which pass the filter.
Each JSON object in the list will give a set of morphologies to fetch, depending on the given
filters.
Note that all attributes are optional, so it’s possible to pass an empty object to fetch all the
morphologies from the database, though it is not recommended.
Create inputs for the Curate, Annotate and Repair workflows¶
The Annotate and Repair workflows should usually be run after the Curate workflow since their inputs should be the outputs of the Curate workflow. But it is still possible to run them on arbitrary inputs (though the morphologies must be valid, as the ones processed by the Curate workflow).
The inputs should consist in:
a directory containing the input morphologies.
a CSV file with the following columns:
morph_path: the path to the morphology file.morph_name: the name of the morphology.mtype: the morphology-type of the morphology (this column is optional).
Any other column is kept into the results but not used in the workflows.
Note that the column names should not contain spaces between commas and names.
a
luigi.cfgfile containing the configuration for all the tasks of the workflow.an optional
logging.conffile containing the logging configuration. If you prefer default logging behavior, remove this file and comment line inlogging_conf_file = logging.confinluigi.cfg.
The examples folder contains
examples for the luigi.cfg and logging.conf files.
These inputs can be created using the Fetch workflow that downloads morphologies from online databases and create the dataset in the correct format.
Run the workflows¶
These workflows are based on the luigi library but can be run via the command line interface.
For example, you can run the Curate workflow with the following command:
morphology-workflows Curate
NOTE: This command must be executed from a directory containing a
luigi.cfgfile. An example of such file is given in theexamplesdirectory.
By default, a local scheduler is used but it is also possible to use a Luigi’s master scheduler
using the -m / --master-scheduler trigger:
morphology-workflows -m Curate
Once the Curate workflow has run, the Annotate and Repair workflows can be run directly, they will just take the results of the Curate workflow as input:
morphology-workflows Annotate
morphology-workflows Repair
Note that it is also possible to run the 3 workflows directly because each workflow depends on each other:
morphology-workflows Repair
More details can be found in the command line interface section of the documentation or by running the commands:
morphology-workflows --help
morphology-workflows <workflow> --help
Results¶
Each workflow will create several new directories, one for each sub-step. These new directories can
be nested into a global result directory for each workflow, using the result_path parameter.
These directories contain intermediate data so it is possible to understand why a morphology
could not be validated at a given step.
The invalid morphologies should be manually fixed before being processed again by the workflows.
The main workflows (Curate, Annotate and Repair) will also create a final CSV file which contains most of the relevant data of the workflow (main sub-step results and final morphology paths). Finally, the Repair workflow can generate morphology releases that contain the final morphologies that could be validated and automatically fixed by the workflow. Usually these morphologies are the most relevant ones for later use.
The morphologies marked as invalid may be fixed manually before rerunning the workflow on them.
Examples¶
The examples folder contains
a simple example that will fetch and process a set of morphologies.
A dataset.csv file is provided which is taken as input for the workflows. A luigi.cfg file
is also provided to give a default configuration for the workflows.
This example can simply be run using the following command:
./run_curation.sh
This script will create a new directory out_curated which will contain the report and all the
results.
Citation¶
When you use this software, we kindly ask you to cite the following DOI:
Funding & Acknowledgment¶
The development of this software was supported by funding to the Blue Brain Project, a research center of the École polytechnique fédérale de Lausanne (EPFL), from the Swiss government’s ETH Board of the Swiss Federal Institutes of Technology.
For license and authors, see LICENSE.txt and AUTHORS.md respectively.
Copyright © 2021-2022 Blue Brain Project/EPFL